We foster an
active
and
innovative
culture within our research group.
Current Research
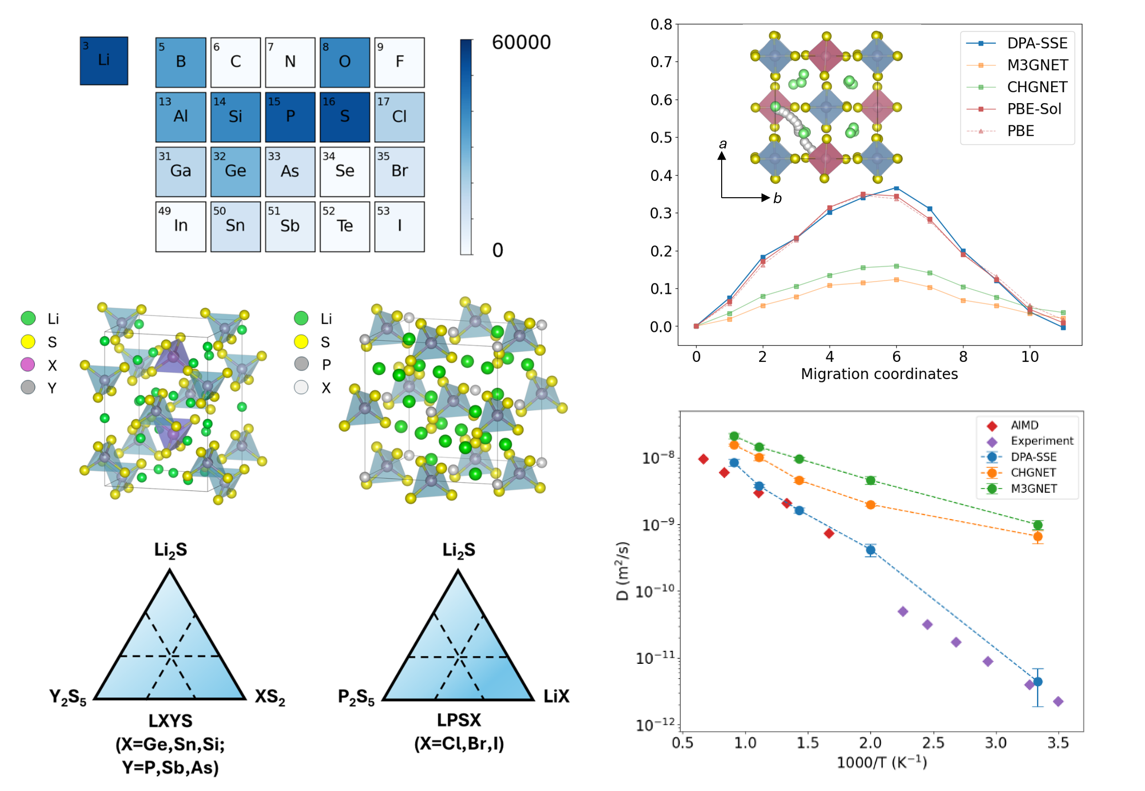
DPA-SSE
This work presents DPA-SSE, a pre-trained deep potential energy model for sulfide-based solid-state electrolytes. The model achieves high accuracy (< 2 meV/atom) while encompassing a broad chemical space, enabling reliable prediction of ionic transport properties. Furthermore, it serves as a foundational model whose fine-tuning and knowledge distillation can significantly accelerate the development of downstream force fields, thereby providing an efficient and robust AI solution for the design of solid-state electrolyte materials.
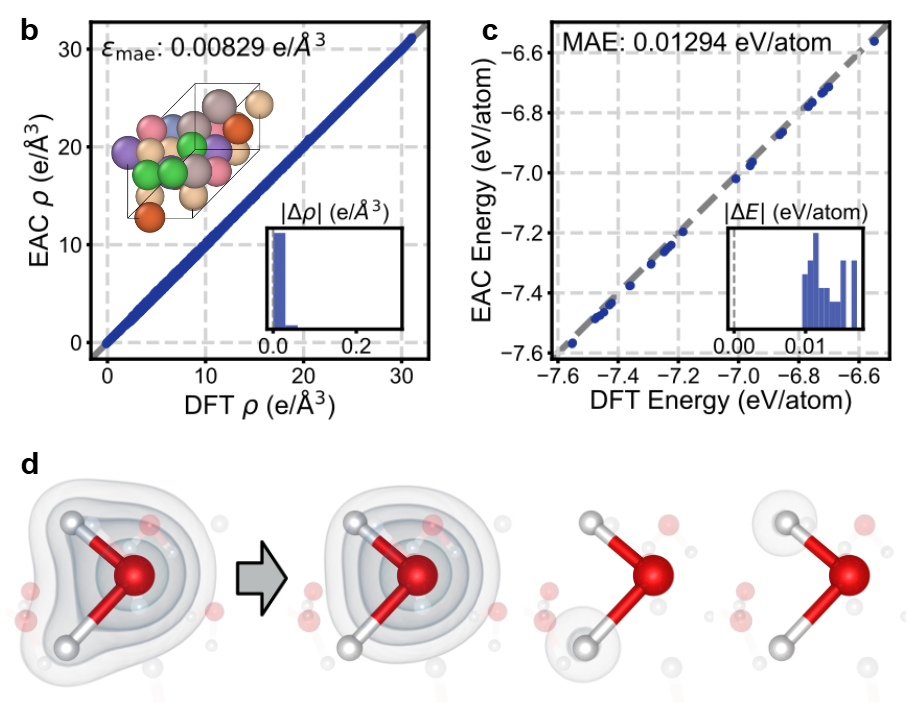
EAC-Net
By extracting atomic contributions that reconstruct the global charge density, our method achieves strong accuracy and scalability for materials containing elements throughout the periodic table.
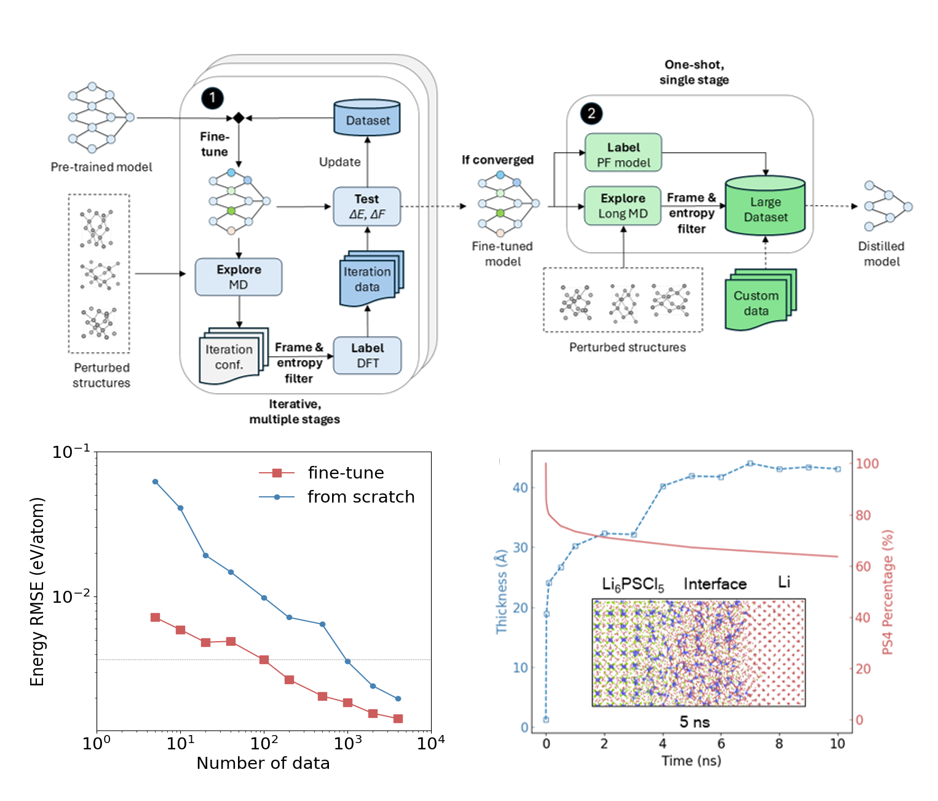
PFD
This work introduces the PFD (Pre-training, fine-tuning, and distillation) workflow, which leverages foundation models to cut DFT data needs by 1–2 orders of magnitude and automatically generate highly accurate and efficient material-specific force fields, enabling atomic simulations of complex systems such as interfaces, amorphous structures, and high-entropy materials.
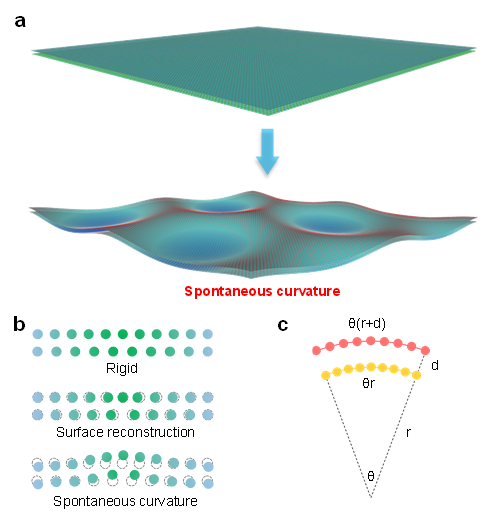
Spontaneous curvature
Based on deep-learning-assisted large-scale molecular dynamics simulations, we unveiled “spontaneous curvature” in two-dimensional heterostructures, driven by the competition between stacking energy and deformation energy. This phenomenon demonstrates robustness in the presence of both thermal fluctuations and interlayer lattice orientation mismatch.